A. Protooncogenes:

Son todos aquellos genes que
participan en los procesos de proliferación e inhibición (factores de
crecimiento, transductores, factores transcripcionales, receptores) siendo
capaces de dar lugar a un oncogen.
Son genes cuyos productos
promueven el crecimiento y la división de la célula. Codifican factores de
transcripción que estimulan la expresión de otros genes, moléculas de
transducción de señales que estimulan la división celular y reguladores del
ciclo celular que hacen que la célula progrese a través de este ciclo.

Cuando un
protooncogén está mutado o se expresa incorrectamente, y contribuye al
desarrollo de un cáncer, pasa a denominarse oncogén que promueve una proliferacion sin control.



Ya sea por mutaciones genéticas (deleción, sumación, transposición o
translocación) del protooncogen o gen supresor tumoral la celula en un dado
momento a convertirse en cancerígena.






Los productos de los
protooncogenes pueden localizarse en la membrana plasmática, en el citoplasma y
en el núcleo celular, y sus actividades se controlan de diversas maneras,
incluyendo la regulación a nivel transcripcional, traduccional y de
modificación de la proteína.
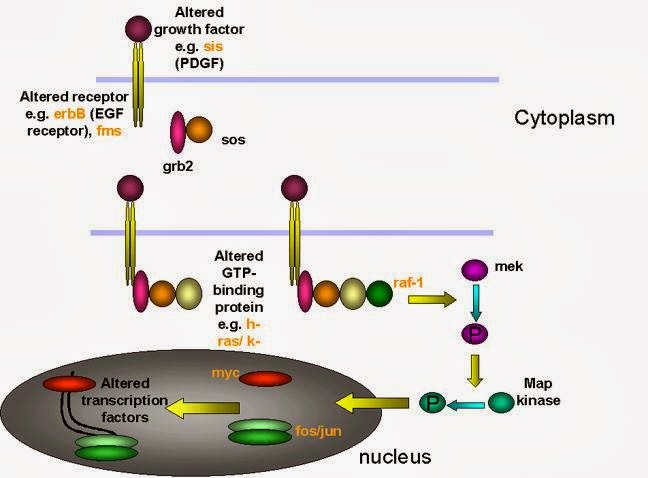
B. Oncogenes
ACTIVACIÓN
Los protooncogenes pueden convertirse en oncogenes mediante uno de tres
mecanismos:
1. Por mutaciones puntuales.
2. Por reordenamientos cromosómicos.
3. Por amplificación de los genes.
MUTACIONES PUNTUALES
Los protooncogenes ras se activan por mutaciones puntuales. Alrededor
del 15 % de todos los tumores humanos tienen oncogenes H-ras o K-ras. Un
posible mecanismo para explicar estas mutaciones es la exposición a sustancias
químicas que producen cáncer.
REORDENAMIENTOS CROMOSÓMICOS
Los reordenamientos cromosómicos parecen activar a los protooncogenes
mediante uno de dos mecanismos:
1. Colocación de los genes en las proximidades de elementos fuertemente
promotores/potenciadores de los loci de receptores de las células T o de
inmunoglobulinas en las células linfoides. En el linfoma de Burkitt, la
translocación t(8;14) coloca al segmento del cromosoma 8 que contiene a c-myc
en estrecha proximidad con el gen de la cadena pesada de inmunoglobulina del
cromosoma 14, cuya expresión es muy activa.
2. Fusión del gen con las nuevas secuencias genéticas. En la leucemia
mieloide crónica, la translocación t(9;22) sitúa al gen c-abl del cromosoma 9
junto al locus bcr del cromosoma 22. El gen híbrido c-abl-bcr codifica una proteína
quimérica que posee actividad de tirosina cinasa. El EWS, otro factor de
transcnpción situado en 22q12, suele encontrarse translocado en muchos
sarcomas, entre ellos el sarcoma de Ewing.
AMPLIFICACIÓN DE LOS GENES
La reduplicación de los protooncogenes puede inducir un aumento de su
expresión o de su actividad. Los ejemplos más representativos son los
siguientes:
1. La amplificación de N-myc en los neuroblastomas;
parece existir una fuerte correlación entre la amplificación de N-myc, el
estadio avanzado y el mal pronóstico.
2.
La amplificación del gen c-erb B2 en el 30 al
40% de los cánceres de mama; existe correlación entre la amplificación de c-erb
B2 y el pronóstico.

C. Genes Supresores







El fracaso de inhibición del crecimiento es
una de las alteraciones fundamentales en el
proceso de carcinógena.
Los Oncogenes dirigen proliferación de
células mientras que los GENES SUPRESORES TUMORALES aplican frenos a la
proliferación celular. Estas forman una red de puntos de control que impiden el
crecimiento incontrolado. Muchos supresores como RB y P53 son parte de una red
reguladora que reconoce la tensión genotoxica de cualquier origen y responde
clausurando la proliferación.
Otro grupo de supresores tumorales está
implicado en la diferenciación celular, haciendo que las células entren en un
fondo común diferenciado posreplicativo posmitotico sin potencial replicativo.
Similar a las señales mitogenas, las señales inhibitorias del crecimiento, se
originan fuera de la célula y usan receptores, transductores de señal y
reguladores de la transcripción nuclear para llevar a cabo sus efectos.
Los productos de los genes supresores
tumorales pueden funcionar como factores de transcripción, inhibidores del
ciclo celular, moléculas de transducción de la señal, receptores de superficie
celular y reguladores de las respuestas celulares al daño del ADN.
RB: Primer y prototipico gen supresor tumoral descubierto, se descubrió
estudiando el retinoblastoma. Producto del gen RB, es una fosfoproteina nuclear
que se expresa de forma obicua y tiene un papel clave en la regulación del
ciclo celular. Existe en estado hipofosforilado activo en las células
quiescentes y en estado Hiperfosforilado inactivo en la transmisión G1/S del
ciclo celular. La importancia de RB se encuentra en su imposición de G1 o el
intervalo entre mitosis y la replicación ADN. En embriones la replicación
comienza después de terminar la mitosis. A medida que continua el desarrollo se
incorpora dos intervalos al ciclo celular: El intervalo 1 entre la mitosis y la
replicación del DN y el intervalo 2 entre la replicación del ADN y la mitosis. (La transición de G1 hasta S es
un punto de control extremadamente importante en el reloj del ciclo celular).
Cruzando el punto g1 pueden pausar el ciclo celular pero están obligadas a terminar
la mitosis. En G1 las celulas pueden salir del ciclo celular, temporalmente QUIESCENCIA o permanentemente SENESCENCIA. G1 se integran diversas
señales para determinar si la célula debe entrar en el ciclo celular, salir del
ciclo celular y diferenciarse o morir.
La replicación de ADN requiere la actividad
de complejos de ciclina E-CDK2 y Ciclina E. Al principio de G1 RB esta activa
fosforilada y se una a los factores de transcripción de la familia E2F
inhibiéndolos, lo que impide la transcripción de ciclina Ey también controla la
estabilidad de inhibidor de ciclo celular p27. RB hipofosforilada bloquea la
transcripción mediada por E2F en dos formas.
- Secuestra E2F impidiendo su interacción con otros activadores de transcripción.
- RB recluta proteínas que remodelan la cromatina (Histona desacetilasa e istona metiltransferasas).
Las señales mitogenas conducen a la
expresión de ciclina D y la activacion de complejos de ciclina D-CDK4/6. Estos
fosforilan RB, inactivando la proteína y liberando E2F para inducir genes diana
como el de ciclina E, esta estimula la replicación de ADN la progresión a través del ciclo celular. Fase s las células están destinadas a
dividirse sin estimulación adicional por factor de crecimiento. Fase M grupos fosfato son eliminados de
RB mediante fosfatasas celulares regenerando la forma hipofosforilada de RB.
Si RB está ausente o capacidad descarrilada
para regular factores de transcripción E2F se liberan los frenos moleculares
del ciclo celular y la célula se desplaza a través del ciclo.
Mutación de RB que en tumores se localizan
en una región de la proteína RB (Bolsillo de RB). Implicada en unión a E2F. La
vía Rb acopla el control de la progresión del ciclo celular en G1 con la
diferenciación lo que puede explicar cómo se asocia la diferenciación con la
salida del ciclo celular.
P53:
Guardian del genoma.
El gen p53 localizado en el cromosoma
17p13.1 es la diana más frecuente para la alteración genética en los tumores
humanos. (TP53 y la proteína p53). 50% de los humanos contiene mutaciones de
este gen. La perdida homocigótica de p53 se encuentra en todos los tipos de
cáncer incluyendo las 3 causas principales de muerte de cáncer (pulmón, colon,
mamas). La herencia de un alelo mutante predispone a los individuos a desarrollar
tumores malignos ya que solo necesitan un solo golpe adicional para inactivar
el segundo alelo normal.
Las mutaciones de p53 sugieren que la
proteína p53 funciona como un guardián critico contra la formación del cáncer.
p53 actúa como un policía molecular que impide la propagación de células
genéticamente dañadas. p53 es un factor de transcripción que está en el centro
de una gran red de señales que detectan tensión celular, como daños del ADN,
telomeros acortados, e hipoxia. Muchas actividades de la proteína p53 están relacionadas
con su función como factor de transcripción. 80% de mutaciones presentes en
canceres humanos se localiza en el dominio de unión al ADN a la proteína. Los
efectos de muchas mutaciones puntuales varían considerablemente; en que algunos
casos existe una abolición completa de las capacidades de transcripción
mientras que otros mutantes conservan la capacidad de unirse a un subgrupo de
genes y activarlos. Mediante mutaciones somáticas y hereditarias las funciones
de p 53 pueden ser inactivadas por otros mecanicismos. Las proteínas transformadoras
de varios virus ADN (E6 de VPH) pueden
unirse a p53 u promover su degradación. La mayoría de tumores sin una mutación
de p53 la función de la vía p53 está bloqueada por una mutación de otro gen que
regula su función.
p53 frustra la transformación neoplasica
mediante tres mecanismos entrelazados:
-
Activación de la detección
transitoria del ciclo celular (Quiescencia)
-
Inducción de una detención
permanente del ciclo celular (Senescencia)
-
Desencadenamiento de la muerte
celular programada (Apoptosis)
Células sanas no sometidas a tensión p53
tiene una vida media corta de 20 minutos debido a su asociación con MDM2.
Cuando la célula está sometida a tensión, p53 sufre modificaciones postranscripcionales
que la liberan de MDM2 y aumenta su vida media. Separada de MDM2, P53 también
llega a activarse como un factor de transcripción.
Genes cuyas transcripción esta
desencadenada por p53 se pueden agrupar en dos categorías: Los que causan
detención del ciclo celular y los que causan apoptosis.
Los iniciadores calve de la vía de
lesión celular son dos proteínas cinasas relacionadas:
-
Mutada de ataxia-telangiectasia
(ATM)
-
Relacionada con la
ataxia-telagiectasia y rad3 (ATR)
La detección de cilco celular mediada
por p53 puede considerarse las respuesta primordial a un daño del ADN. Fase G1
causada principalmente por transcripción dependiente de p53 del inhibidor de
CDK CDKN1A (p21). P21 inhibe los complejos ciclina-CDK y los foforilacion de
RB, impidiendo que las células entren en fase G1.
P53 también ayuda en el proceso mediante
la inducción de ciertas proteínas como GADD45 que ayudan a la reparación de
ADN. P53 puede estimulas las vías de reparación del ADN mediante mecanismos
independientes de la transcripción. Si el daño se repara con éxito p53 regula
positivamente la transcripción de MDM2 conduciendo a su propia destrucción,
liberando el bloque celular. Si la lesión no puede reparase la células puede entrar
en senescencia inducida por p53 p sufrir apoptosis dirigida por p53.
La senescencia inducida por p53 es una
detención permanente del ciclo celular caracterizada por cambios específicos en
la morfología y la expresión génica. Los mecanismos de la senescencia incluyen
cambios epigeneticos que dan lugar a la formación de heterocromatina en
diferentes loci en todo el genoma. Estos focos de heterocromatina en diferentes
asociados a la senescencia incluyen permanentemente la expresión de estas
dianas de E2F.
La apoptosis inducida por p53 de las
células con daño irreversible del ADN es el mecanismo protector final contra la
transformación neoplasica. P53 dirige la transcripción de varios genes
proapoptosicos como BAX y PUMA. La afinidad de p53 por los promotores e
intensificadores de los genes de reparación de ADN es más fuerte que su
afinidad que por los genes
proapoptosicos. Primero se estimula la vía de reparación de ADN y finalmente si
el daño del ADN no se repara se acumula suficiente p53 para estimular la
transcripción de los genes proapotosicos y la célula muere.
VIAS DE LAS APC /B-
CATENINA
• Genes de la poliposis
adenomatosa del colon- genes supresores- regulación negativa de señales que
provueven el crecimiento.
• Mutaciones en APC-
poliposis adenomatosa familiar-adolecentes o tercera edad.
• Polipos sufren
transformación maligna –cáncer de colon-fuerte predisposición hereditaria
• Función importante de APC
regular negativamente a B catenina ya que en auncensia de WNT, APC degrada B
catenibna evitando acumulacion en citoplasma.
• APC: regulacion de la
estabilidad y funsion de la B catenina
• APC y B catenina
son componentes de la via de señal WNT
• Celulas en reposo – no
expuestas a WNT- complejo B catenina-APC- destruccion de B catenina.
• Expuestas a WNT- no
destruccion de B catenina y concentracion citoplasmatica aumenta.
• B catenina se transloca al
nucleo se une a TCF –activa genes de la
progresion del ciclo celular.
• APC mutado o ausente no destruccion de B catenina y sucede igual
como que WNT estubiera presente.
INK4aARF
DOS
PRODUCTOS PREOTEICOS:
- CDKI p16/INK4a que
bloquea la fosforilacion de RB.
- P14/ARF activa la via
p53 al inhibir a MDM” e inpedir la destruccion de p53.
Ø
La
mutacion impacta en RB y p53 y promover tumores en vegiga, cabeza , cuello,
leucemias linfoblasticas agudas y colangioscarcinomas ademas cancer cervical.
VIA TGF-B
TGF-B
potente inhibidor de la proliferación de
células epiteliales, endoteliales y hematopoyéticas normales.
Regula
mediante la unión a un complejo serina-treonina cinasa-receptores TGF-B I y II,
esto conduce a la activación de cinasa y fosforilacion de R-SMAD (traductor de
señales anti proliferativas) los cuales
pueden entrar en el núcleo y unirse a SMAD4
y activar la trascripción de genes CDKI p21 y p15/INK4b y además TGF-B
conduce a la represión de c-MYC, CDK2, CDK4 y ciclinas A y C.
Estos
cambios dan lugar a una disminución en
la fosforilación de RB y a la detención del ciclo celular.
Cáncer
los efectos inhibitorios de TGF-B están afectadas por mutaciones que afectan al
receptor II ( cáncer de estomago, colon y endometrio)
La
inactivación mutacional de SMAD4 cáncer pancreático.
PTEN
• Homologo de fosfatasa y
tensina asociada a la membrana, codificada por el gen del cromosoma 10q23 que
esta mutado en e síndrome de Cowden (crecimientos benignos, como tumores de los
apéndices cutáneos, canceres epiteliales, de mama y endometrio.
• PTEN actúa como supresor tumoral
al servir como freno en la via de supervivencia y el crecimiento de PI3K/AKT.
• PI3K (puede estar mutado
incrementando la señal) fosforila el lípido inositidocinasa-inositido 3, 4,
5-trifosfato que se une a la cinasa PDKI y la activa-fosforila y activa a AKT
mediante una serie de sustratos BAD y MDM2 e intensifica la supervivencia
celular además inactiva TSC1/TSC2 que estan mutados en esclerosis tuberosa.
• La inactividad de TSC1/TSC2
activa a mTOR que estimula la captación de nutrientes (glucosa y aa)
• La perdida adquirida de
PTEN es una de las vías mas frecuentes por las que se regula positivamente la
señal PI3K/AKT en varios canceres.
NF1
La
herencia de un alelo mutante de este gen provoca neurofibromas benignos
numerosos y gliomas del nervio óptico (neurofibromatosis tipo 1)
Algunos
de ellos se desarrollan a malignos.
Neurofibromina
es producto proteico de NF1 con dominio activador GTPasa que regula la
transducción de señal a través de la proteína RAS ( que transmite señales
promotoras de crecimiento y oscila entre estadios de unión a GDP inactivos como
GTP activos.
La
neurofibromina convierte RAS desde activo a inactivo con la perdida de neurofibromina RAS queda
activo.
NF2
• las mutaciones en estos
genes predisponen a neurofibromatosis tipo 2, las mutaciones en este gen
desarrollan schwanomas benignos bilaterales del nervio acústico, si las
mutaciones afectan ambos alelos se
pueden encontrar meningionas y ependimomas.
• El producto de este gen se
llama neurofibromina 2 o merlina (
miembro clave de la vía supresora tumoral).
• Las vías de señales
controla el tamaño orgánico mediante la modulación del crecimiento, la
proliferación y la apoptosis celular.
VHL
Las
mutaciones del gen 3p se asocian a canceres de células renales,
feocromocitomas, hemangioblastomas del SNC, angiomas retinianos, quistes
renales hereditarios.
Es
parte de un complejo ubicutin ligasa un sustrato critico para esta actividad es
el factor de transcripción inducido por hipoxia ( HIF1a).
En
presencia de O2, HIF1a es hidroxilado y se une a la proteína VHL condiciendo a
la ubicutinizacion y degeneración proteosomica.
En
ausencia de O2, HIF1a escapa del reconocimiento de VHL y la consiguiente
degradación.
HIF1a
puede translocarse al núcleo y poner en marcha muchos genes como VEGF y PDGF.
La
ausencia de VHL impide la ubicutinizacion y degradación de HIF1a y se asocia
con niveles aumentados de factores de crecimiento angiogénicos.
WT1
Localizado
en el cromosoma 11p13 se asocia con el
desarrollo del tumor de Wilms, un cáncer de riñón pediátrico.
La
proteína WT1 es un activador de la transcripción de genes implicados en la
diferenciación renal y gonadal.
Regula
la transición mesenquimatoso a epitelial
que tiene lugar en el desarrollo del riñón.
El
efecto oncogéno de la deficiencia de WT1 esta íntimamente conectado con el
papel del gen en la diferenciación de los tejidos genitourinarios.
PTCH
PTCH
1 y 2 son genes supresores tumorales que codifican una proteína de membrana
celular (PATCHED) que funciona como receptor para una familia de proteínas
llamadas hedgehog que regula varios genes como TGF-B, PDGFRA y PDGFRB.
Las
mutaciones se relacionan con el síndrome
de Gorlin ( carcinoma de las células basales nevoide. Causado por la exposición
de rayos UV.
Referencias
bibliográficas
- Kumar, Vinay; Abbas, Abul K; et al. Patología estructural y funcional. Robbins y Cotran. 8va. edición, año 2010. Editorial Elsevier, España, S.A. Capítulo 7. Págs. 278-296.
- Bruce, Alberts. Biología Molecular. Ediciones Omega. Barcelona, 1996
- Karp Gerald. Biologia Celular y Molecular. 2006
- Brandon N , Juaristi J, Aguirre V , Romero Benítez M. Universidad del noreste de Argentina, Facultad de Ciencias Médicas, Catedra de Bioquímica. Oncogenes y Genes supresores de tumores.
- Brandan, Nora Profesora Titular. Cátedra de Bioquímica. Facultad de Medicina. UNNE. Juaristi, Julián Profesor Titular. Cátedra de Bioquímica. Carrera de Enfermería. Facultad de Medicina. UNNE. Aguirre, Victoria Profesora Adjunta. Cátedra de Bioquímica. Facultad de Medicina. UNNE. Romero Benítez, Margarita Jefa de Trabajos Prácticos. Cátedra de Bioquímica.Universidad Nacional del Nordeste Argentina, Facultad de Medicina, ONCOGENES Y GENES SUPRESORES DE TUMORES, Pags. 5,6.
excelente muy entendible
ResponderBorrar