Mientras los oncogenes dirigen la proliferación de las células, los productos de los genes supresores tumorales aplican frenos a la proliferación celular. Los productos proteicos de los genes supresores tumorales pueden funcionar como factores de transcripción, inhibidores del ciclo celular, moléculas de transducción de la señal, receptores de superficie celular y reguladores de las respuestas celulares al daño de ADN.
RB
La proteína RB, el producto del gen RB, es una fosfoproteína nuclear que se expresa de forma ubicua y tiene papel clave en la regulación del ciclo celular. RB existe en estado hipofosforilado activo en las células quiescentes y en estado hiperfosforilado inactivo en la transición G1/S del ciclo celular. La importancia de RB se encuentra en su imposición de G1, o el intervalo entre la mitosis (M) y la replicación del ADN. Una vez que las células cruzan el punto de control G1, sin embargo, las células pueden pausar el ciclo celular por un tiempo, pero están obligadas a completar la mitosis. En G1, sin embargo, las células pueden salir del ciclo celular, bien temporalmente, lo que se llama quiescencia o bien permanentemente, llamada senescencia.
La iniciación de la replicación del ADN requiere la actividad de complejos de ciclina E-CDK2 y la expresión de la ciclina E depende de factores de transcripción de la familia E2F. Al principio de G1, RB está en su forma activa hipofosforilada y se une a los factores de transcripción de la familia E2F inhibiéndolos, lo que impide la transcripción de ciclina E. RB hipofosforilada bloquea la transcripción mediada por E2F al menos de dos formas. En primer lugar, secuestra E2F impidiendo su interacción con otros activadores de la transcripción. En segundo lugar, RB recluta proteínas que remodelan la cromatina. Estas enzimas modifican la cromatina de modo que hacen los promotores insensibles a los factores de transcripción.
Si RB está ausente (como resultado de mutaciones génicas) o su capacidad para regular los factores de transcripción E2F está descarrilada, se liberan los frenos moleculares del ciclo celular y la célula se desplaza a través del ciclo. Por tanto, la vía RB acopla el control de la progresión del ciclo celular en G1, con la diferenciación, lo que puede explicar cómo se asocia la diferenciación con la salida del ciclo celular.
P53: guardián del genoma
 |
| Papel de RB en la regulación del punto de control G1-S del ciclo celular. RB hipofosforilado en el complejo con los factores de transcripción E2F se une al ADN, recluta factores de remodelación de la cromatina e inhibe la transcripción de genes cuyos productos se requieren para la fase S del ciclo celular. Cuando RB es fosforilado por los complejos ciclina D-CDK4 y ciclina E-CDK2, libera E2F. Entonces este último activa la transcripción de genes de fase S. |
El gen p53 se localiza en el cromosoma 17p13.1 y es la diana más frecuente para la alteración genética en tumores humanos.
El hecho de que las mutaciones de p53 sean frecuentes en diversos tumores humanos sugiere que la proteína p53 funciona como un guardián crítico contra la formación del cáncer. En efecto, es evidente que p53 actúa como un “policía molecular” que impide la propagación de células genéticamente dañadas. P53 es un factor de tanscripción que está en el centro de una gran red de señales que detectan tensión celular, como daños del ADN, telómeros acortados e hipoxia. Muchas actividades de la proteína p53 están relacionadas con su función como factor de transcripción
Igual que sucede con RB, las proteínas transformadoras de varios virus ADN, incluyendo las proteínas transformadoras de varios virus ADN, incluyendo la proteína E6 del VPH, pueden unirse a p53 y promover su degradación. También, de forma análoga a RB, se piensa que en la mayoría de tumores sin una mutación de p53 la función de la vía p53 está bloqueada por una mutación de otro gen que regula su función.
P53 frustra la transformación neoplásica mediante tres mecanismos entrelazados: activación de la detención transitoria del ciclo celular (quiescencia), inducción de una detención permanente del ciclo celular (senescencia) o desencadenamiento de la muerte celular programada (apoptosis)
Se han encontrado cientos de genes cuya transcripción está desencadenada por p53. Pueden agruparse en dos categoría amplias: los que causan detención del ciclo celular y los que causan apoptosis. Si el daño del ADN puede repararse durante la detención del ciclo celular, la célula revierte a su estado normal; si la reparación fracasa, p53 induce apoptosis o senescencia.
Se ha demostrado que p53 activa la transcripción de la familia mir34 de los ARNm (mir34a-mir34c). Los ARNm se unen a secuencias en la región no traducida 3´de los ARNm, impidiendo la traducción. La regulación de p53 por los mir34 explica, al menos en parte, cómo p53 es capaz de reprimir la expresión génica, y parece que la regulación de este ARNmi es crucial para la respuesta a p53.
Vía de la APC/β-catenina
 |
| A. Papel de p53 en el mantenimiento de la integridad del genoma. B. p53 media la represión génica mediante activación de la transcripción de los ARNmi. p53 activa la transcripción de la familia mir34 de ARNmi. Los mir34 reprimen la traducción tanto de genes proliferativos, como las ciclinas, como de genes antiapoptósicos como BCL2. La represión de estos genes puede promover la quiescencia o bien la senescencia, así como la apoptosis. |
Vía de la APC/β-catenina
Los genes de la poliposis adenotamosa del colon (APC) tiene como principal fucion la regualcion negativa de señales que promueven el crecimiento, la mutacion de estos se asocia a al poliposis adenomatosa familiar que desarrollan pólipos adenomatosos en el colon. Estos pólipos pueden sufirir una transformación maligna dando lugar a un tumor. Una función importante de la proteína APC es regular negativamente a la β-catenina.
En el nucleo la β-catenina forma un complejo con TCF, ciclina D1 y otros genes.
La importancia de la vía de señal APC β-catenina en la oncogenia se da por el hecho de que los tumores de colon que tiene genes APC normales albergan mutaciones del gen de β catenina que impiden su destrucción por APC.
INK4a/ARF
Este locus codifica dos proteínas que son CDKIp16/INK4a que bloquea la fosforilacion de RB mediada por ciclina D/CDK2. El otro es p14/ARF que activa la via p53 al inhibir a MDM2 e impedir la destrucción de p53.
La via TGF-β
TGF-β es un potente inhibidor de la proliferación mediante la unión a un complejo serina-treonima cinasa. la unión al receptor conduce a la activación de la cinasa y la fosforilacion de receptores SMAD. Los R-SMAD puede entrar en el núcleo unirse a SMAD-4 y activar la transcripción de genes como CDKIp21 y p15/INK4b. este gen también conduce a al represión de c-MYC, CDK2, CDK4 y ciclinas A y E, estos cambios producen una disminución de la fosforilacion de RB y la detención del ciclo celular.
PTEN
Es una fosfatasa asociada a la membrana codificada por un gen del cromososma 10q23, este gen esta mutado en el síndrome de Cowden que se caracteriza por el crecimiento benigno como tumores de apéndices cutáneos. PTEN actua como supresor tumoral al frenar la via promotora de la supervivencia y el crecimiento de PI3K/AKT.
NF1
Este gen produce la neurofibromina que contiene un dominio activador de GTPasa que regula la transducción de la señal a través de RAS (transmite señales promotoras del crecimiento). La neurofibromina facilita la conversión de RAS de un estado activo a un estado inactivo.
VHL
La proteína VHL es parte de un complejo ubicuitina ligasa. En presencia de oxigeno la HIFIα se une a la proteína VHL llevando a su degradación. En estados hipoxicos esta escapa de VHL y se transloca al nucleo y pone en marcha mucos genes como los factores de crecimiento/angiogenicos, VEGF y PDGF.
WT1
La proteína WT1 activa la transcripción de genes para la diferenciación renal y gonadal. El efecto oncogeno de la deficiencia de WT1 Está conectado con el papel del gen en la diferenciación de los tejidos genitourinarios.
Patched (PTCH)
PTCH! Y PTCH2 codifican una proteína llamada PATCHED que funciona como receptor de las proteínas Hedgehog, esta vía regula varios genes como TGF-β y PDGFRA.
Patogenia del Retinoblastoma
Según la hipótesisis propuesta por Knudson:
• Se requieren dos mutaciones que afecten ambos alelos RB, en el locos 13q14 para desarrollar un retinoblastoma.
• Los niños heredan una copia defecetiva del gen RB, mientras que la otra copia es normal. Se presenta el retinoblastoma cuando el alelo RB normal está mutado en los retinoblastos, como consecuencia de una mutación somática espontánea.
• En casos esporádicos, ambos alelos RB deben sufrir mutación somática en el mismo retinoblasto.
El cáncer se desarrolla cuando la célula pierde la heterocigosidad para el gen RB normal.
Aproximadamente el 60% de los retinoblastomas son esporádicos, y el resto son familiares, transmitiéndose la predisposición a desarrollar el tumor como un rasgo autosómico dominante. El 40% de los casos se dan en individuos que heredan una mutación germinal con un alelo RB.
Los portadores de un mutante del gen supresor tumoral del Rb tienen un riesgo 10.000 veces mayor de desarrollar retinoblastoma.
La proteína RB, es el producto del gen Rb, tiene un importancia e la regulación del ciclo celular. Su imposición se encuentra en G1, el intervalo entre la mitosis y la replicación del ADN. En G1 las células pueden salir del ciclo celular (quiescencia), o hacerse permanente (senescencia).
En la replicación del ADN se necesita la actividad de complejos de cidina E-CDK2, la expresión de ciclina E, depende de los factores de transcripción de la familia E2F. La proteína RB, secuestra E2F impidiendo su interacción con otros activadores de trancripción. RB recluta proteínas que remodelan la cromatina, que se unen a los genes que responden a promotores de E2F como la ciclina E, que modifican la cromatina que hacen a los promotores insensibles a los factores de transcripción.
Los miembros de la familia Rb pueden complementar parcialmente su función en tipos celulares diferentes de los retinoblastos, por medio de proteínas bolsillo, que se unen a los factores de transcripción E2F. Estas proteínas regulan la progresión a través del ciclo celular.
La pérdida de control del ciclo celular normal es central para la transformación maligna, y que al menos de cuatro reguladores clave del ciclo celular aparecen desregulados en la inmensa mayoría de los cánceres humanos.

El retinoblastoma es un cáncer del ojo que se origina en la retina, la capa de células sensibles a la luz que recubren la parte posterior del ojo y convierte los rayos de luz en impulsos eléctrico

El retinoblastoma es
un tumor maligno de la
retina que afecta,
generalmente a niños menores de 6
años de edad. El tumor se puede localizar en uno o en ambos ojos y,
normalmente en nuestro medio, se diagnostica antes de que se extienda a otras
partes del cuerpo.
Cuando el retinoblastoma es hereditario (40 % de los casos) la primera mutación se hereda de uno de los padres y la segunda se produce durante el desarrollo de la retina. En caso de padres afectos de la enfermedad existe alrededor de un 50 % de posibilidades de que los hijos tengan la mutación y un alto porcentaje de que desarrollen retinoblastoma.
Síntomas:
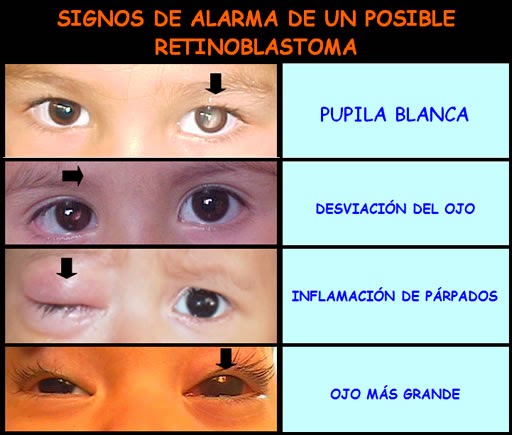
Uno o ambos ojos pueden resultar afectados.
La pupila puede aparecer blanca o tener manchas blancas. Con frecuencia, se observa un brillo blanco en el ojo en las fotografías tomadas con flash. En lugar del típico "ojo rojo" del flash, la pupila puede parecer blanca o distorsionada.
Otros síntomas pueden abarcar:
• Estrabismo convergente
• Visión doble
• Ojos desalineados
• Enrojecimiento y dolor en el ojo
• Visión deficiente
• Iris que puede ser de diferente color en cada ojo
Si el cáncer se ha diseminado, se puede presentar dolor óseo y otros síntomas.



Síntomas:
Uno o ambos ojos pueden resultar afectados.
La pupila puede aparecer blanca o tener manchas blancas. Con frecuencia, se observa un brillo blanco en el ojo en las fotografías tomadas con flash. En lugar del típico "ojo rojo" del flash, la pupila puede parecer blanca o distorsionada.
Otros síntomas pueden abarcar:
• Estrabismo convergente
• Visión doble
• Ojos desalineados
• Enrojecimiento y dolor en el ojo
• Visión deficiente
• Iris que puede ser de diferente color en cada ojo
Si el cáncer se ha diseminado, se puede presentar dolor óseo y otros síntomas.
Calcificación
Bibliografía:
- Dome JS, Rodriguez-Galindo C, Spunt SL, Santana VM. Pediatric sold tumors. In: Abeloff MD, Armitage JO, Niederhuber JE, Kastan MB, McKenna WG, eds. Abeloff's Clinical Oncology. 4th ed. Philadelphia, Pa: Elsevier Churchill Livingstone; 2008:chap 99.
- Zage PE, Herzog CE. Retinoblastoma. In: Kliegman RM, Behrman RE, Jenson HB, Stanton BF, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, Pa: Saunders Elsevier; 2011:chap 496.
No hay comentarios.:
Publicar un comentario